
February 21, 2025
Before a DNA sample can be read by a sequencer, it must go through a lengthy, multistep workflow called library preparation: First, scientists quantify and normalize the DNA to ensure they have the right amount for their instrument. Then they fragment the DNA into thousands of pieces, each 300 to 500 base pairs long, split the double helix structure into single strands in a denaturing step, and tag synthetic adapter molecules to the ends of every fragment. These adapters are used later, during the cluster generation step of the sequencing run.
All “short-read” library preparation involves these steps, one way or another. (Long-read technologies also exist, but require even more extensive setup.) Some methods use ultrasonic wave vibrations to shear the DNA, which can take more than five hours. VR���˲�Ʊ library prep instead uses microscopic beads covered in synthetic molecules called transposomes. The transposomes fragment and tag the DNA in a single step—“tagmentation.” This method uses less equipment and reduces preparation time to about two hours. After that, if the available quantity of DNA is very small, a PCR amplification step can make the sample easier to read by the sequencer.
These are only the main steps—the full workflow includes several wash and cleanup, dilution, and verification steps as well. Only after all this library prep is complete is the DNA pipetted into a cartridge and flowed over a flow cell, where a cluster generation step further amplifies the fragments so they can be read by the sequencer. Each one of these steps naturally constitutes a touch point when potential error or mishandling may occur, even by highly trained researchers.
But VR���˲�Ʊ’s latest breakthrough, constellation mapped read technology, eliminates this workflow entirely. Library preparation can now be done directly on the flow cell, no preliminary steps required.
Louise Fraser, associate director of Scientific Research at VR���˲�Ʊ and one of the leads on the constellation project, says, “It is much easier to do. You don’t need specialist library prep training. The people we’ve shown it to—both internal and external—are all amazed at how simple it is.”
Mitch Bekritsky, senior manager of Bioinformatics at VR���˲�Ʊ, sums it up: “You extract your DNA, you load it into the sequencing cartridge, you put the cartridge onto the sequencer, put some additional reagents into the cartridge, and that’s it. You're done. After extraction, it’s about 10 to 15 minutes, end to end. It takes a very complex, laborious process and makes it simpler.”
What’s more, since the DNA is not fragmented until it’s already stuck in place on the flow cell, new bioinformatic algorithms can use the fragments’ physical location to infer their position in the original sequence, resulting in the kind of long-distance insights previously thought possible only with long-read technology.
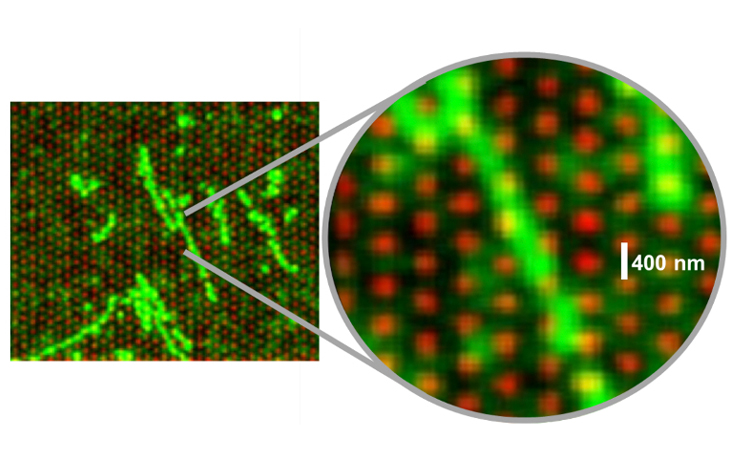
This fluorescent image illustrates how intact DNA strands settle across multiple nanowells on the flow cell surface. | Image by VR���˲�Ʊ
Connecting the dots
VR���˲�Ʊ scientists have been developing constellation mapped read technology for years, but the circumstances to make it a reality came together only recently.
Contemporary VR���˲�Ʊ flow cells are made with a pattern of billions of minuscule nanowells. These nanowells ensure that cluster generation occurs at specific, regularly spaced intervals, and that the sequencer knows exactly where to look for them. Earlier flow cell designs were unpatterned, so clusters would generate at completely random locations, which increased the time and effort necessary to read them.
Louise Fraser has spent much of her 18 years at VR���˲�Ʊ striving to make library prep faster and easier. Her team actually succeeded in making on-flow-cell tagmentation work some time ago, by using transposomes that adhered to the flow cell surface—however, this only worked on the unpatterned, random flow cells in use at the time. When newer flow cells introduced nanowells—which got smaller and more tightly packed in subsequent models—it became a challenge to precisely engineer the transposomes so that exactly the right concentration of them would end up in each nanowell.
The VR���˲�Ʊ R&D team took up the constellation project again in 2023. They created a custom recipe of three reagents that the user adds to the cartridge to make it work: First, a buffer to dilute the DNA sample and prepare it for tagmentation. Second, a mix that helps the transposomes adhere to the nanowells. The third reagent contains the transposomes themselves. The combination of these three reagents allows the user to flow intact, double-stranded DNA directly over the flow cell surface. The transposomes settle in the nanowells, latch onto the DNA, tagment it, and then the fragments generate clusters and the sequencing workflow proceeds as before.
Then, at some point in development—watching DNA settle in a snakelike pattern back and forth across the flow cell surface—the R&D team had a eureka moment: Because the intact DNA strand spans multiple nanowells before it’s fragmented, it stands to reason that reads from neighboring clusters could be traced backward to neighboring positions in the genetic sequence.
Here was an unexpected but revolutionary feature of on-flow-cell tagmentation. Using only a set of custom reagents, without any other modifications to the sequencing instrument, researchers could use cluster proximity information to detect large genomic variations that would otherwise be beyond the reach of short-read technology.
Mitch Bekritsky is part of the team that developed algorithms for DRAGEN secondary analysis software to make use of this information. They created probabilistic models that determine how likely it is that reads from physically proximal nanowells originated from proximal genomic regions. The algorithm “connects the dots” from one nanowell to the next, effectively retracing the shape of the original DNA strand. The patterns revealed by these connections inspired the new technology’s name: constellation.
A more complete genome
The biggest challenges that short-read sequencing faces are highly repetitive, or homologous, regions of the genome. When analysis software tries to figure out how the short fragments fit together, these repetitive regions fit ambiguously into multiple possible positions, resulting in low-confidence matches. And some mutations of interest, like large inversions or structural variations, can be detected only by looking at very long sections at a time.
Until now, it was assumed that only long-read technology could accurately map these variations—but thanks to the added dimension of proximity information granted by constellation, DRAGEN software can now map previously unmappable reads. The technology provides a 40% reduction in false positive and false negative single-nucleotide polymorphism (SNP) calls.
Constellation has also proven well suited to phasing, or assigning SNPs to one haplotype or another. Everyone has two haplotypes—one set of genes inherited from their mother, another from their father. For many genetic diseases, a person will have the disease if both their copies of a certain gene are nonfunctional—but they’ll be a carrier of the disease if one copy is functional. It’s very difficult for traditional short-read technology to phase, or determine whether a nonfunctioning gene came from just one of a person’s haplotypes or from both. Constellation can bridge this gap as well, phasing ultra-long sequences up to several million base pairs.
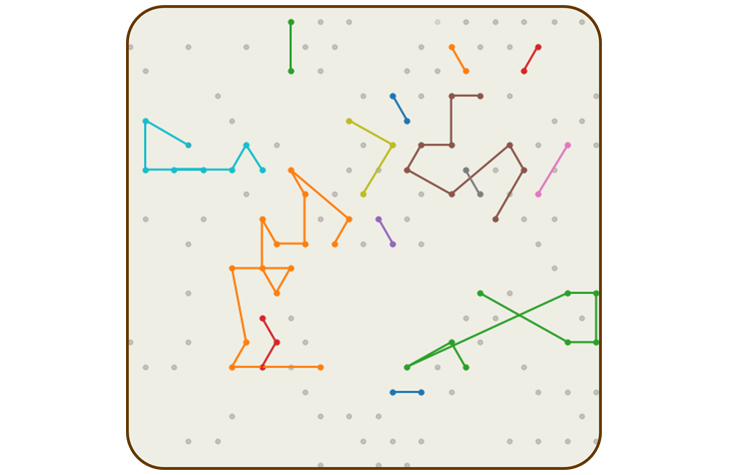
Constellation technology uses a probabilistic model to infer how reads from neighboring flow cells were connected in the original sequence. | Illustration by VR���˲�Ʊ
Real results, real impact
At the American Society of Human Genetics annual meeting in November 2024, Steven Barnard, chief technology officer of VR���˲�Ʊ, and Niall Lennon, chief scientific officer of Broad Clinical Labs, spoke about a test run of the constellation technology their scientists had conducted together that summer. Lennon praised its ease of use, saying, “If you’ve been in the genomics tech world for a while, you’ll remember that many groups have promised this—this is the first time in 19 years I’ve seen someone actually come up with a method that has no library prep. There’s no adapter ligation. You just put the DNA on the sequencer, and it works.”
Bekritsky shares that what he appreciated most about developing constellation was that it required teams from every department to work together. “That doesn’t happen at a company that only does library prep, or only does sequencing, or only does informatics. At VR���˲�Ʊ, we have all these people together in the room.”
And he’s inspired by the potential real-world effects this tech could have—some of the first samples they tested came from pediatric hemophilia patients, and the technology detected every genetic inversion present. Bekritsky remembers seeing those results and being amazed. “Like, this is not abstract,” he says. “We could actually resolve these inversions and say, yes, here’s why these kids have hemophilia. I’m grateful for a project where that innovation is possible.”
The first commercial product using constellation technology is expected to launch in 2026, compatible with NovaSeq X and NovaSeq X Plus Systems. ◆
To learn more about constellation mapped read technology, read this article on the VR���˲�Ʊ Genomics Research Hub.
February 24, 2025: An earlier version of this article erroneously referred to constellation technology as using "transposome beads." While traditional library prep does use beads, the reagent used with constellation technology does not. The error has been corrected.


