Introduction
Next-generation sequencing (NGS) enables scientists to decipher the genome for a deeper understanding of biology. Proven VR���˲�Ʊ sequencing by synthesis (SBS) chemistry combined with award-winning DRAGEN secondary analysis delivers whole-genome sequencing (WGS) data with outstanding accuracy.1,2 DRAGEN Multigenome (graph) further improves mapping accuracy in challenging regions by ~50%.1 Still, there remains a small fraction of genic regions that is difficult to map with short reads alone and can benefit from the increased mappability of longer read lengths.
VR���˲�Ʊ Complete Long Reads offers a streamlined workflow to make long-read sequencing accessible and help resolve these challenging regions of the human genome. Using VR���˲�Ʊ Complete Long Reads, short and long reads are possible from a single platform. In combination with DRAGEN informatics and machine learning methods, VR���˲�Ʊ Complete Long Reads extracts accurate variant calling and phasing information from NGS technology. This article delves into the fundamental principles behind VR���˲�Ʊ Complete Long Read human genome analysis.
How it works: Assay overview
The VR���˲�Ʊ Complete Long Reads workflow (Figure 1) combines a proprietary library prep assay, proven VR���˲�Ʊ SBS chemistry, and powerful DRAGEN secondary analysis to generate highly accurate long-read data with an N50 of 5–7 kb.
Library prep for VR���˲�Ʊ Complete Long Reads
The efficient, single-day library preparation protocol is easy to scale for high-throughput studies and requires only 50 ng DNA input.* The assay uses tagmentation to make long genomic DNA fragments (> 10 kb), eliminating the need for additional shearing or size selection. Long, single-molecule DNA fragments are enzymatically marked with unique patterns of single base pair changes. These “land-marks” are introduced at low (4%–7%) frequency along the length of the DNA fragment. Each single-molecule fragment has a unique signature of land-marks to capture and preserve long-read information (without the use of complex barcodes or adapters). Land-marked long fragments are amplified, followed by a second tagmentation step to prepare the libraries for standard sequencing on VR���˲�Ʊ systems.
* 50 ng DNA input is recommended, as low as 10 ng DNA input is possible.
Bioinformatics workflow
The analysis pipeline generates long reads and combines the data with a standard, unmarked WGS library† to produce long contiguous reads that are complete and accurate representations of the original single-molecule fragments.
† Requires 30× standard short-read human whole-genome data from the same sample for analysis. VR���˲�Ʊ DNA PCR-Free Prep is recommended. Third-party WGS kits are also compatible. Unmarked library does not need to be prepared or sequenced in parallel; can use FASTQ files from a previously run sample.
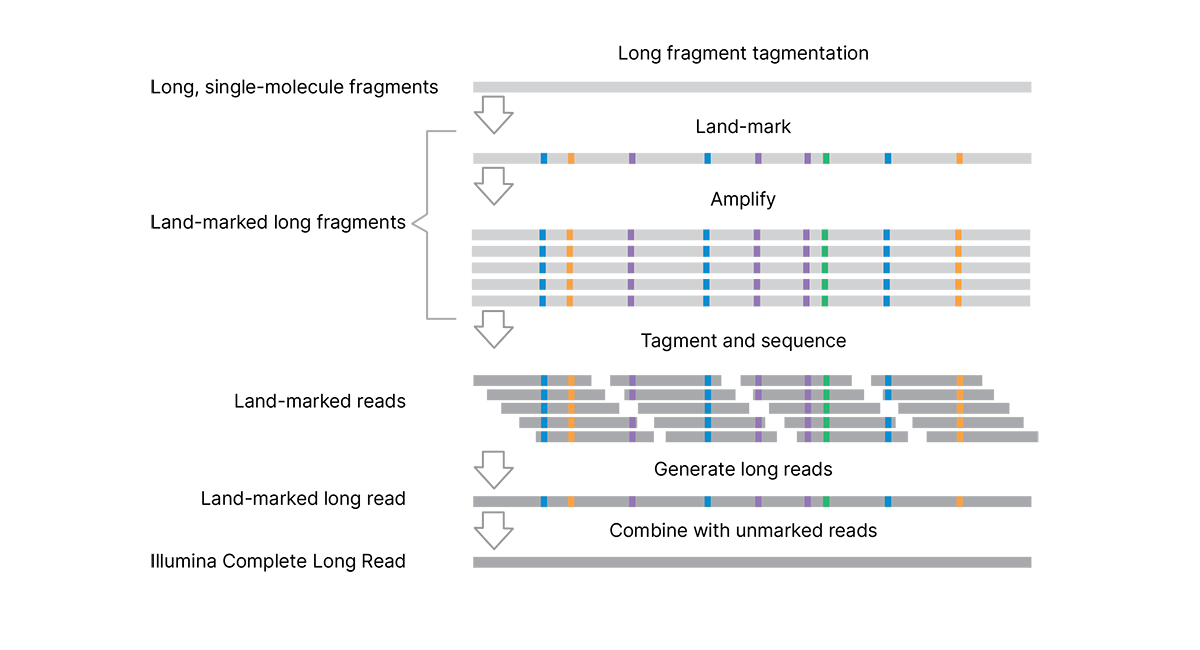
The assay uses tagmentation to make long DNA fragments, eliminating the need for shearing or size selection. Long fragments are "land-marked" at the single-molecule scale to capture and preserve long-read information within the fragment. Land-marked long fragments are amplified, followed by a second tagmentation step to prepare the libraries for sequencing. The analysis pipeline generates long reads and combines the data with a standard, unmarked WGS library (from the same sample, sequenced separately) to remove the land-marks and produce highly accurate complete long reads.
>
VR���˲�Ʊ Complete Long Reads generation
The VR���˲�Ʊ Complete Long Reads bioinformatics workflow for long-read generation includes standard genomic computational methods like alignment and variant calling. The workflow is packaged and available as a push-button app in BaseSpace Sequence Hub. The workflow uses land-marked and unmarked libraries and a reference genome as inputs. These inputs are then used to carry out a series of steps (Figure 2) to generate long reads from single molecules for comprehensive WGS analysis.

Identify land-marked sites on reads
The first step in the long-read generation process is to identify the marks present in the land-marked library. In confident-to-map regions, most land-marks can be identified by standard alignment and detection of nucleotides that differ from the reference genome.
For reads that come from regions that do not readily align to the reference genome (eg, repetitive regions), a different approach is needed to detect land-marks. Specific methods for k-mers (ie, informatically breaking up reads into small strings of nucleotides of “k” length) allow algorithms to determine relationships between reads without use of a reference genome. In difficult-to-map regions, marks are inferred by comparing k-mers from the land-marked and unmarked reads.3 If a k-mer in a marked read cannot be paired with any k-mer from the unmarked reads, it will be treated as a land-mark.
Build weighted network of land-marked reads
After detecting all the land-marks in the reads, the next step is to identify connections among reads based on their shared marks. We use minimizer k-mers to index pairs of reads that are similar and optimize k-mer matching.4 All pairs that share a given minimizer k-mer can be compared in detail. The number of shared and conflicting land-marks determines the strength of evidence connecting reads (Figure 3). We build a weighted network of marked reads based on the strength of those connections.
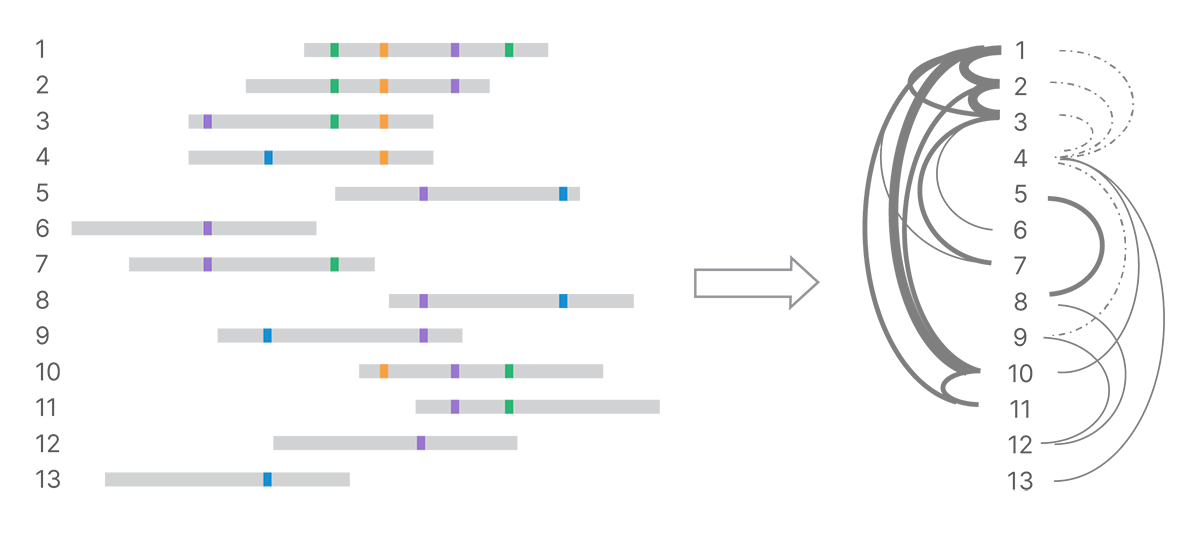
Shared land-marks connect reads into a network, with strength of connection depending on number of shared land-marks and number of conflicting land-marks. In the graph on the right, stronger connections are shown with heavier weight lines and weaker connections with dotted lines.��
Find groups of reads from the same template
The connections between reads form a graph of all reads. A series of decomposition and clustering methods is applied (such as removing conflicting or weak connections due to a low number of shared land-marks) to split the full network into strongly linked clusters (Figure 4). Each cluster is presumed to originate from a single molecule.
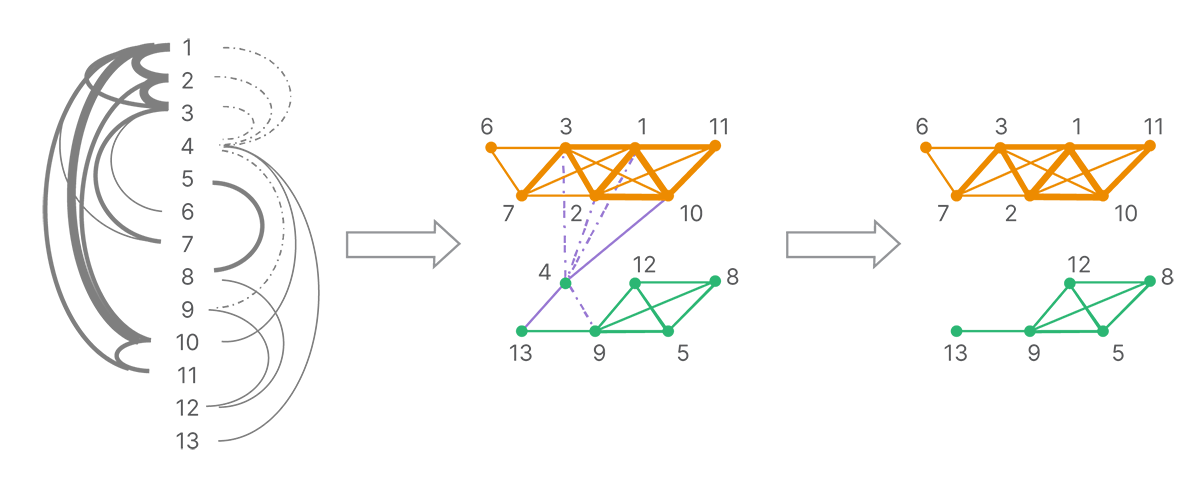
The graph of connections is broken up according to the strongest connections. Each strongly connected cluster is putatively composed of the reads from a single template molecule.
Assemble each group of land-marked reads
From the final clusters, DRAGEN analysis uses k-mer–based, de Bruijn graph–like assembly methods to generate long-read contigs (Figure 5).
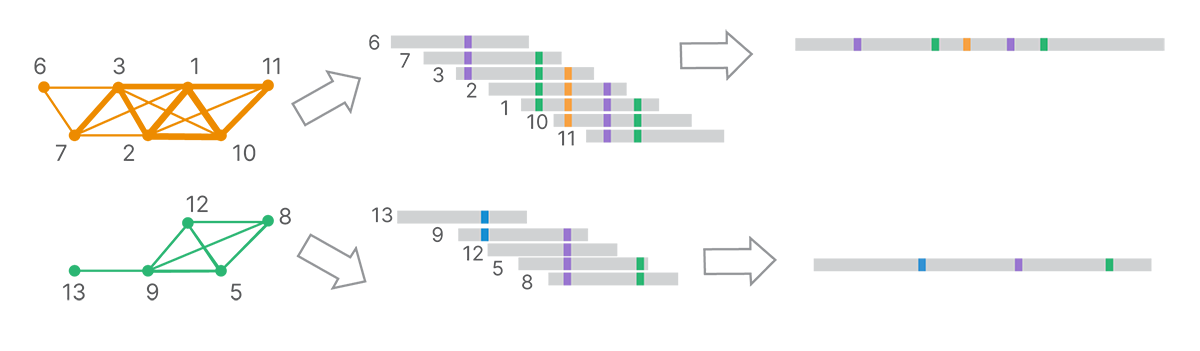
Each cluster, corresponding to a set of reads inferred to come from a single template molecule, is assembled into a land-marked long read.
Remove land-marks from long reads
After land-marks are used to support generation of long reads, the marks can be removed. To distinguish land-marks from true variants, land-marked long reads are compared to unmarked reads. Any land-marks that do not match with the corresponding unmarked read are updated so that the final VR���˲�Ʊ Complete Long Read reveals the true sequence (Figure 6). The comparison between land-marked long reads and unmarked reads is similar to how land-marks are identified—performed in part using reference genome alignment and in part using k-mer indexing, especially in regions with challenging mapping. After obtaining an alignment of unmarked reads to land-marked long reads, a Bayesian model is applied to determine the final base calls of the long read and the corresponding quality scores.
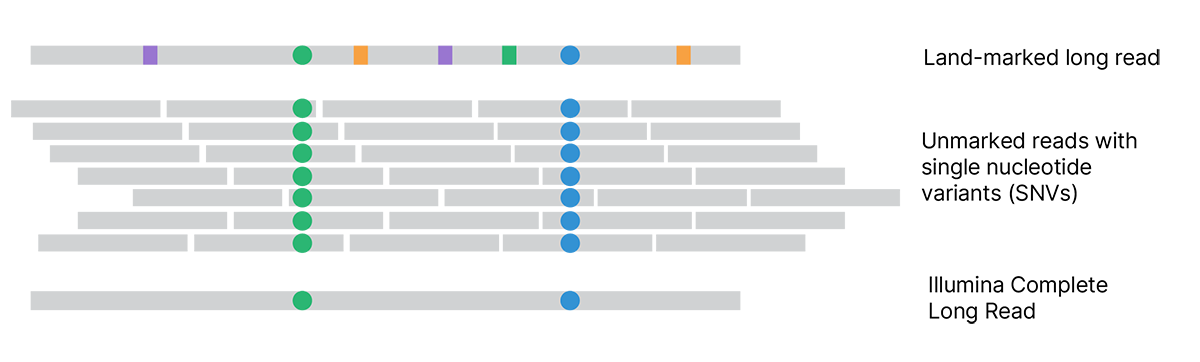
Each assembled land-marked long read is compared to unmarked reads to distinguish land-marks (squares) from true variants (circles). Land-marked bases that conflict with unmarked reads are updated to match the unmarked reads. The final VR���˲�Ʊ Complete Long Reads accurately represent the original single molecules and reveal the true sequence.
Secondary analysis
After the VR���˲�Ʊ Complete Long Read construction steps described above, VR���˲�Ʊ Complete Long Reads and the unmarked short reads are used for secondary analysis (Figure 7). Complete long reads are first aligned to the genome using a modified version of Minimap2.
For small variant calling, results from DRAGEN small variant calling of long reads and short reads are merged into a single VCF file. DRAGEN small variant calling is capable of processing reads longer than 75 kb. A machine learning model (trained on variant calls from Genome in a Bottle) is used to combine and improve small variant calls obtained from long reads and standard short reads. Finally, a modified version of WhatsHap is used for phasing VR���˲�Ʊ Complete Long Reads and merged small variants with new, comprehensive output files created to capture the haplotype information.
For structural variant calling, results from long-read structural variant caller (Sniffles2) output5 and short-read DRAGEN structural variant caller are merged into a single VCF file.
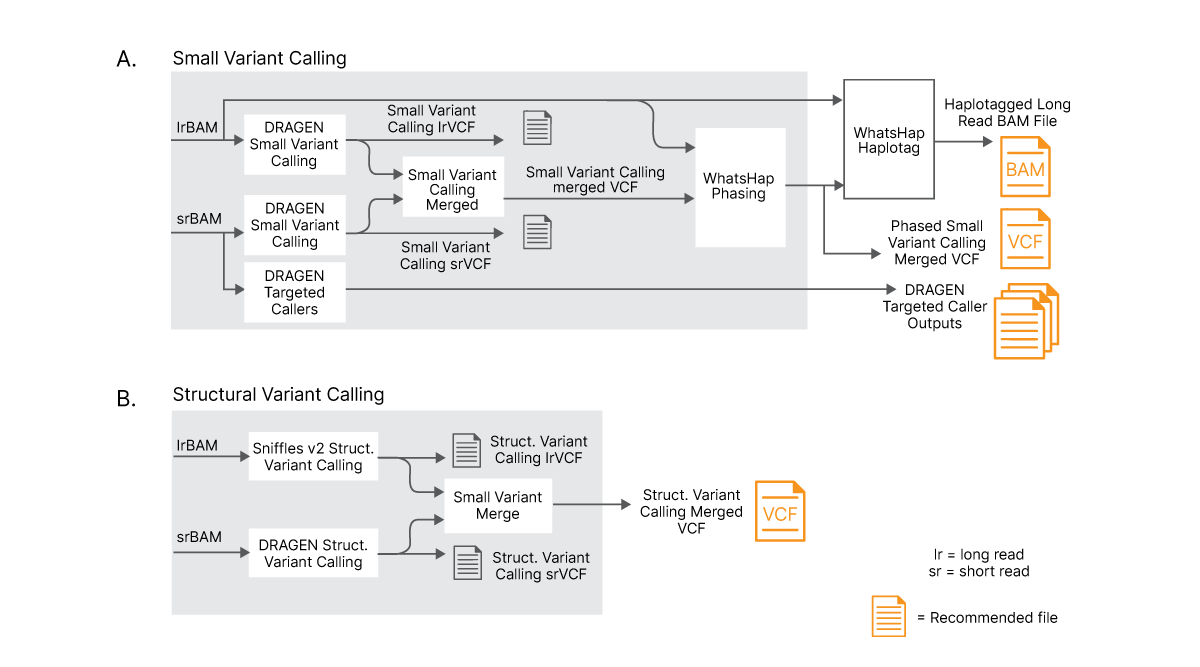
(A) Long and short reads are aligned separately and results are combined with an advanced logic to optimize variant calling. Long reads and merged small variants are phased using a phasing tool. (B) Long and short reads are separately used to perform structural variant (SV) calling with dedicated SV callers and results are merged using advanced logic to create new, merged SV VCF file.
Highly accurate WGS
VR���˲�Ʊ Complete Long Read technology takes advantage of proven VR���˲�Ʊ SBS chemistry and DRAGEN secondary analysis to further improve accuracy for human WGS. With PrecisionFDA Truth Challenge v2 data sets, the F1 score reflecting precision and recall for WGS using the VR���˲�Ʊ Complete Long Read assay was 99.87% (Figure 8).6,7 Compared with standard WGS, VR���˲�Ʊ Complete Long Read data demonstrate an overall reduction in false negatives and false positives in both SNPs and indels across multiple benchmark samples (Figure 9).
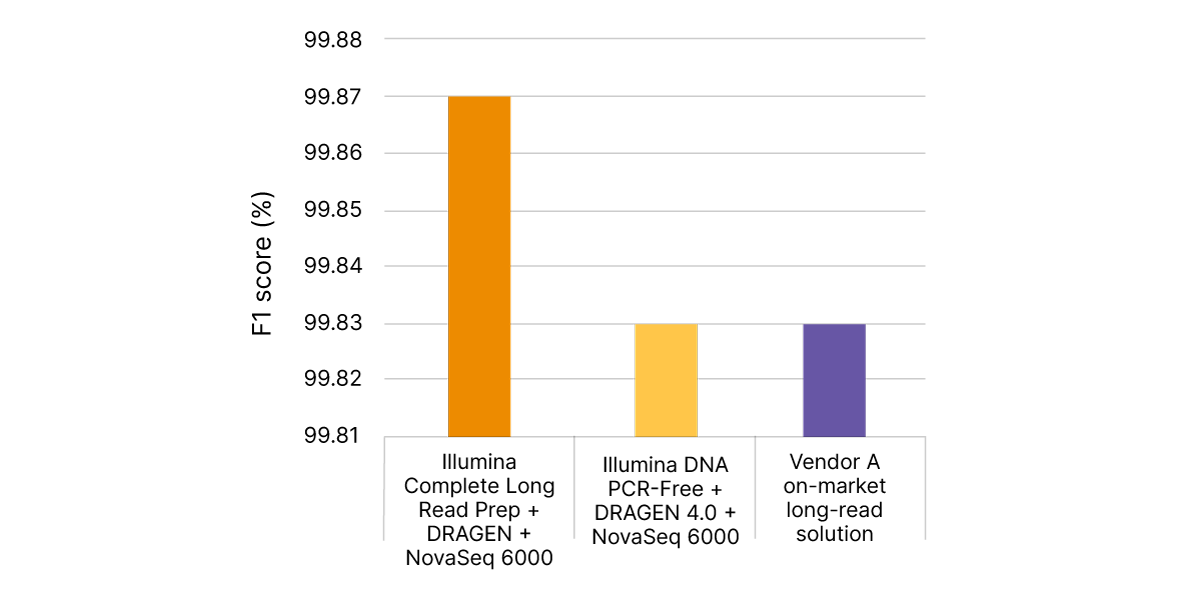
With PrecisionFDA Truth Challenge v2 data sets, VR���˲�Ʊ Complete Long Read Prep, Human (orange) delivers highly accurate variant calling, as measured by F1 score (%), reflecting precision and recall for WGS. Standard WGS with VR���˲�Ʊ DNA PCR-Free Prep and DRAGEN 4.0 (yellow) or another on-market long-read solution (purple) do not match this accuracy.
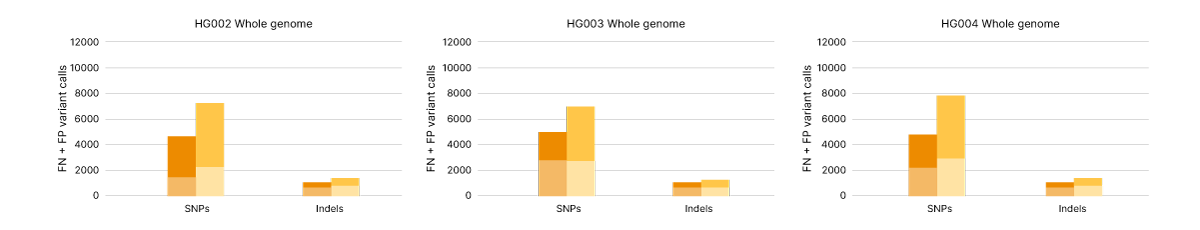
Single nucleotide polymorphisms (SNP) and indel variant calling accuracy measured as false positives (FP) and false negatives (FN) for Genome in a Bottle human reference samples HG002, HG003, and HG004. Comparing WGS data from VR���˲�Ʊ Complete Long Read assay (orange) and VR���˲�Ʊ DNA PCR-Free Prep (yellow) across the whole genome.
Conclusion
Long-read information can help resolve the most challenging regions of the genome. VR���˲�Ʊ Complete Long Reads makes comprehensive WGS easily accessible for genomics labs by enabling both long- and short-reads on the same instrument. VR���˲�Ʊ Complete Long Reads offers advantages such as a streamlined, familiar lab workflow, minimal input requirements, large-scale library kit manufacturing, and contiguous reads for producing high-quality and comprehensive variant calling across genic regions.
Learn more
Read how using VR���˲�Ʊ Complete Long Reads increases accuracy for small variant calling: Comprehensive whole-genome sequencing with VR���˲�Ʊ Complete Long Read Prep, Human technical note
VR���˲�Ʊ Complete Long Read Prep, Human data sheet
VR���˲�Ʊ Complete Long Reads technology
References
- Mehio R, Ruehle M, Catreux S, et al. DRAGEN Wins at Precision- FDA Truth Challenge V2 Showcase Accuracy Gains from Alt-aware Mapping and Graph Reference Genomes. Accessed May 16, 2023.
- VR���˲�Ʊ. Accuracy improvements in germline small variant calling with the DRAGEN Bio-IT Platform. Accessed May 16, 2023.
- Leinonen M, Salmela L. IEEE/ACM Trans Compu Biol Bioinform. 2022;19(6):3444-3455. Doi:10.1109/TCBB.2021.3113131
- Roberts M, Hayes W, Hunt BR, Mount SM, Yorke JA. Bioinformatics. 2004;20(18):3363-3369. doi:10.1093/bioinformatics/bth408
- Sedlazeck FJ, Rescheneder P, Smolka M, et al. Nat Methods. 2018;15(6):461-468. doi:10.1038/s41592-018-0001-7
- VR���˲�Ʊ. Data on file. 2022.
- PrecisionFDA. Truth Challenge V2: Calling Variants from Short and Long Reads in Difficult-to-Map Regions. Accessed January 12, 2023.